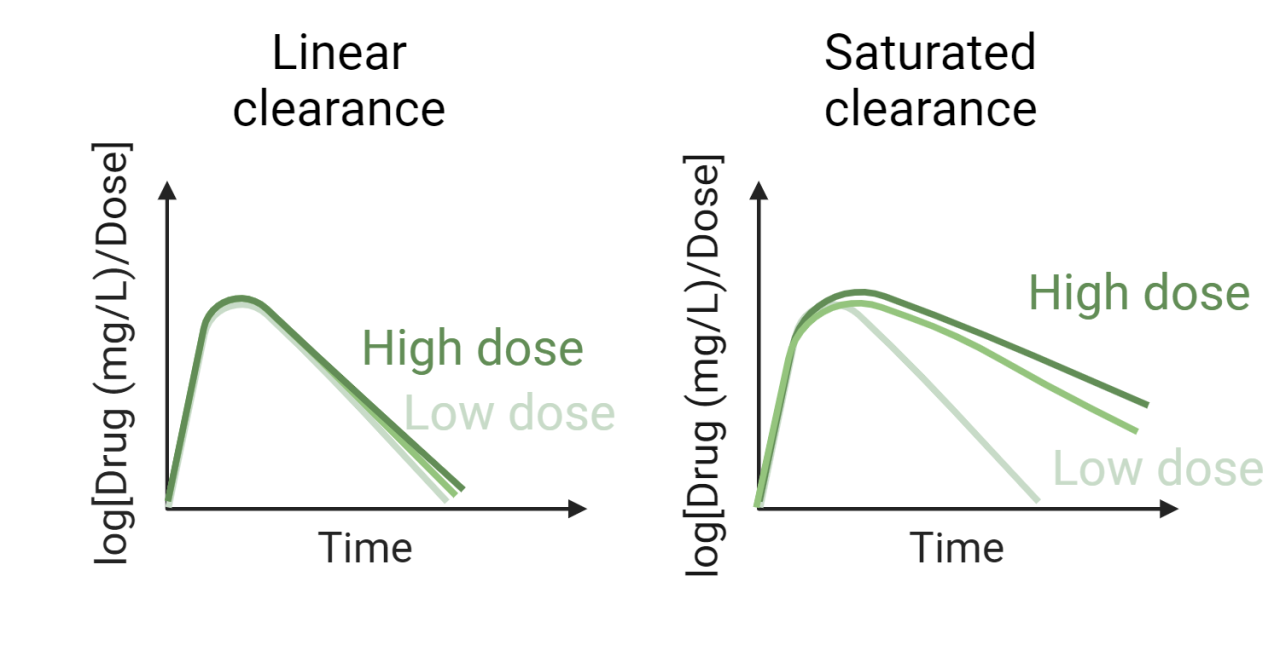
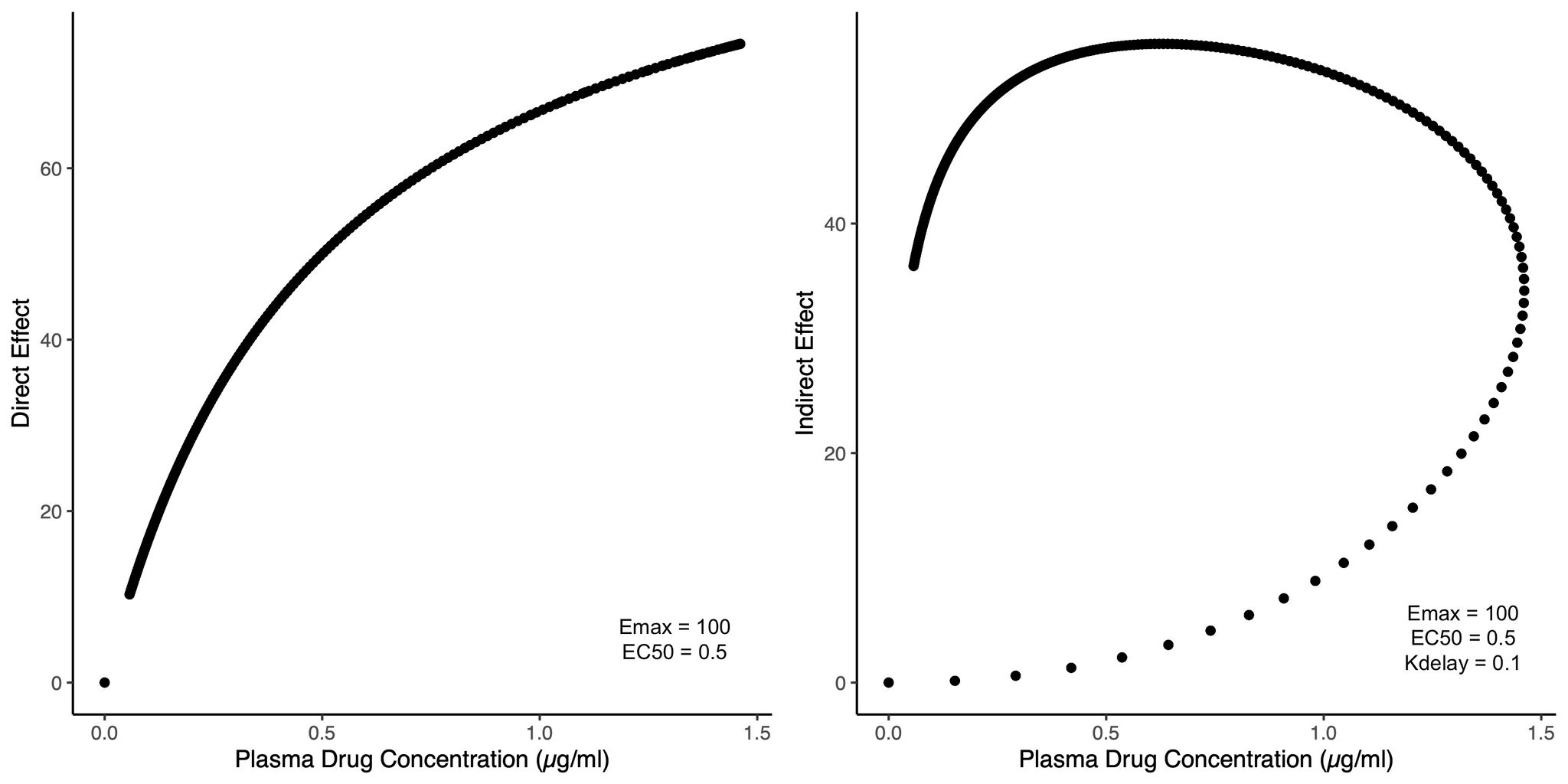
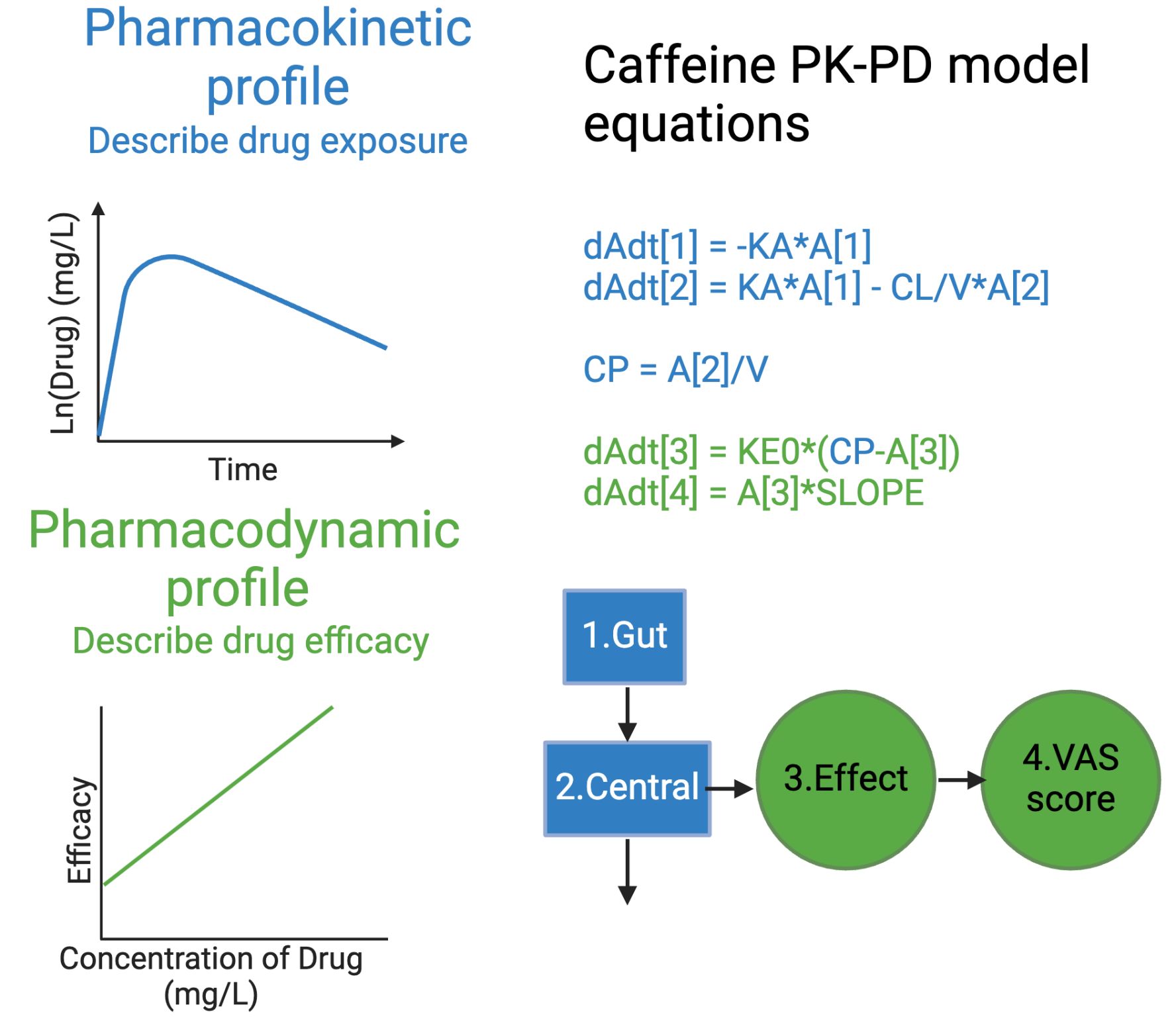
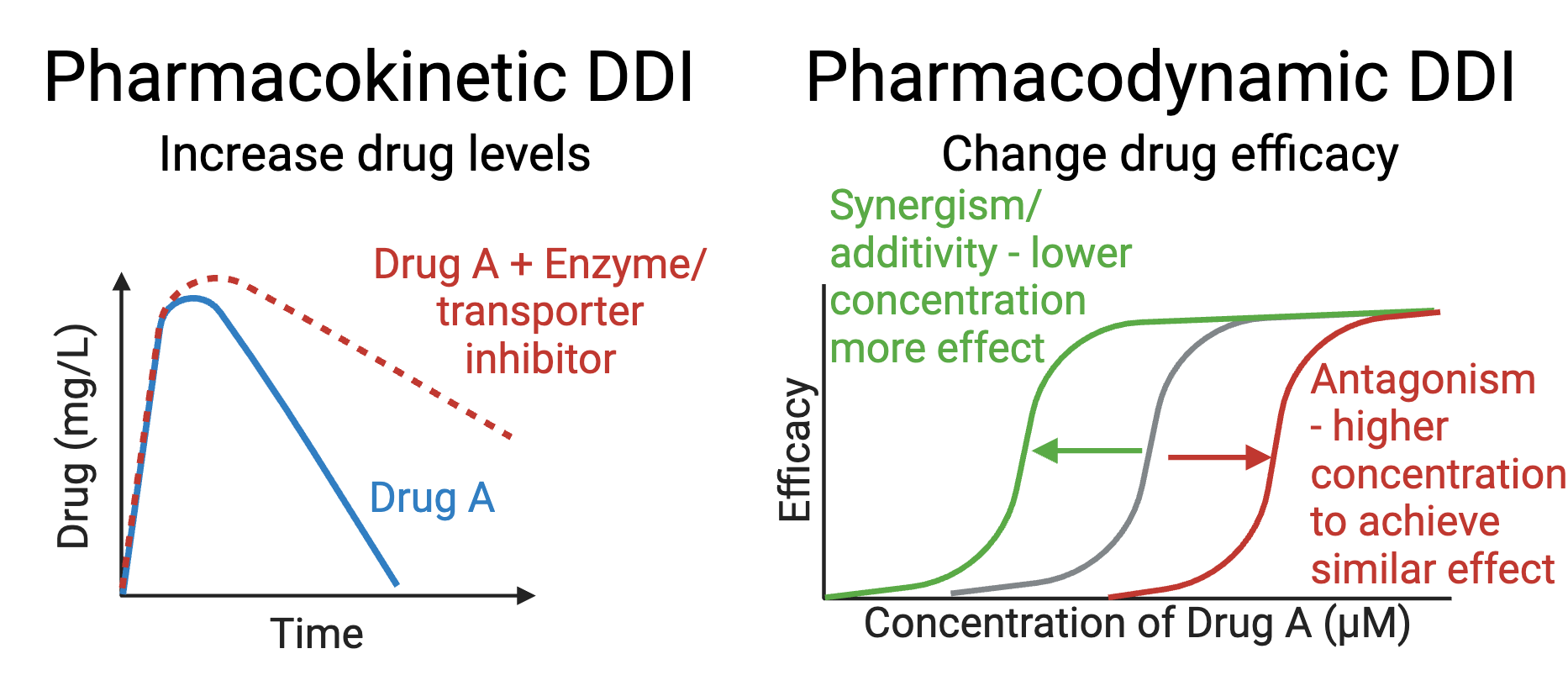
As a (mostly) computational scientist, I was excited to see both the Nobel prizes in physics and chemistry being awarded for computational work last week. In particular, the Physics price on Artificial neural networks, a fundamental building block in many artificial intelligence (AI) tools, was an exciting recognition. As the nobel prize website aptly puts it, artificial neural nets help us to independently discover properties in data by using neural net structures to process information. This opens up the possibility of discovering more trends e.g. non linear ones, that classical machine learning methods might struggle to work with. Allowing AI to handle the job further allows us to analyze large amounts of big data, which would have previously been impossible for humans to manually go through and analyze ourselves. It is thus no wonder AI has such a pervasive use today.
What about the use of AI in pharmacometrics?
Much interest has come about in recent years merging pharmacometrics tools with AI. In literature today, tools have been developed promising to automate pharmacokinetic (PK) model building and prediction of new dosing regimens, integrate unstructured data as covariates in models, and even provide mathematical equations describing previously unknown relationships between trends, to name a few.
Does this mean that I am now out of a job and should pursue my grad school fever dream of opening a bakery or becoming a tik tok creator though?
Not really.
As British statistician George Box famously said, “All models are wrong, but some are useful.” Regardless of whether a model is being built by me or by an AI model, stringent evaluation of a model on its usefulness still applies, and findings that come out of these analyses still need to be critically evaluated using a scientific lens. And, as many science papers have concluded, further work and experimental evaluation always need to be done to fully confirm our hypothesis.
AI tools are thus unlikely to completely take over our jobs. Rather, they should be seen as an upgrade to complement traditional pharmacometrics tools, allowing us to work more efficiently and make even more exciting discoveries.